Remove duplicate computation of divergence function #7
Conversation
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
Sign up for free
to join this conversation on GitHub.
Already have an account?
Sign in to comment
1 participant
Add this suggestion to a batch that can be applied as a single commit.
This suggestion is invalid because no changes were made to the code.
Suggestions cannot be applied while the pull request is closed.
Suggestions cannot be applied while viewing a subset of changes.
Only one suggestion per line can be applied in a batch.
Add this suggestion to a batch that can be applied as a single commit.
Applying suggestions on deleted lines is not supported.
You must change the existing code in this line in order to create a valid suggestion.
Outdated suggestions cannot be applied.
This suggestion has been applied or marked resolved.
Suggestions cannot be applied from pending reviews.
Suggestions cannot be applied on multi-line comments.
Suggestions cannot be applied while the pull request is queued to merge.
Suggestion cannot be applied right now. Please check back later.
Hello, I've been working with AlphaBeta on a large dataset for Prof. Johannes, which is split up into > 100 separate windows. Because it takes quite a while to run AlphaBeta for each window, I looked at your code to find some quick opportunities for speedups. I found this:
Which can be improved to this:
Because this function is a key part of the optimization loop, it is time critical -> The second version leads to a 2x speed up.
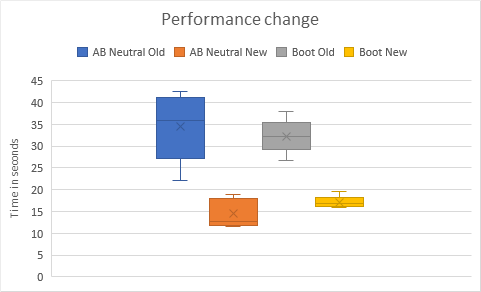
The change is applicable to AB Neutral & the BOOT model (so 4x in total) but also for all the other models.
I calculated the speedup with the
AlphaBetafile underVignettesand the librarymicrobenchmark(Code below)Do you know of any other simple speedups? I'm happy to implement them :)
BTW, thanks for the library, it's really useful!
Benchmark code